
Author: Dustin Newman
Most clients send us a vial, capsule, pouch, powder, tablet, or finished product expecting the report to tell them whether the lot is acceptable.
Here’s what is often overlooked: even the most accurate laboratory can only test what you actually submit. If that sample doesn’t truly represent the batch, shipment, container, or population you care about, the report can be technically correct and still miss the real quality question.
Sampling is more than paperwork. It operates as an integrated component within the testing process.
At Vanguard Laboratory, we work with manufacturers, supplement brands, food companies, contract packagers, distributors, importers, and resellers of specialty ingredients and research peptides. The sampling strategy doesn’t have to be complicated, but it does need to match the actual risk. One vial may be enough to answer, “What’s in this vial?” It’s rarely enough to answer, “Is the entire lot acceptable?”
FDA and USP answer different questions
FDA and USP are often mentioned together, but they don’t do the same job.
FDA requirements focus on whether your quality system has scientifically sound, documented sampling and testing controls. For finished pharmaceuticals, 21 CFR 211.160 requires laboratory controls to include scientifically sound specifications, standards, sampling plans, and test procedures, with samples that are representative and properly identified. 21 CFR 211.165 requires appropriate laboratory determination of conformance before release and states that written sampling and testing plans must include the method of sampling and the number of units per batch to be tested when such plans are used.
USP, by contrast, tells you what an official article must meet when tested by a compendial procedure. The USP General Notices state clearly that conclusions about compendial conformance apply to the units tested, and that the compendium does not prescribe the frequency of batch testing or the extrapolation of results. Passing a USP method on the one sample submitted proves exactly that: the sample tested met the criteria. It does not automatically prove the whole lot meets specification.
That distinction is important. FDA looks at the overall quality system and the decision-making process. USP defines the performance standard for the article that was actually tested. Many good release, verification, and supplier-qualification decisions need both: the right analytical method and the right sampling plan.
One good sample can still create false confidence
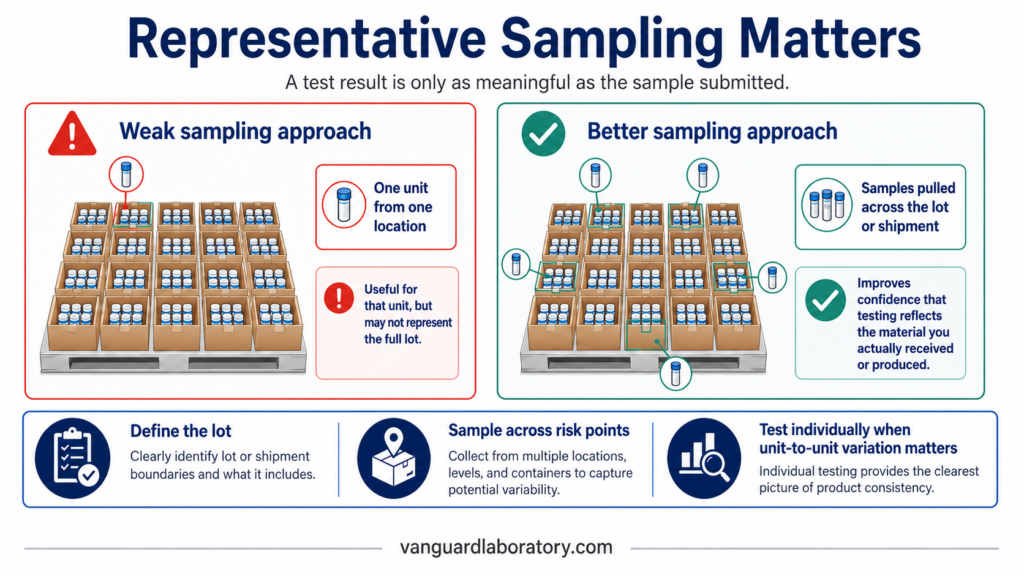
Real products vary. Powders segregate. Capsules reflect blend inconsistencies. Peptide vials can differ in fill volume, concentration, residual moisture, vial position, or lyophilization behavior. Finished goods can be affected by labeling mix-ups, supplier substitutions, storage conditions, packaging errors, or shipment damage.
Assuming independent random selection from a large lot, if a defect affects 1% of the lot, testing one unit gives you only about a 1% chance of catching it (calculated as 1 − (0.99)^n). Three units raise that to roughly 3%. Ten units get you to about 9.6%. That doesn’t mean every project needs hundreds of samples. It means one clean report should never be treated as automatic proof of lot uniformity unless the sampling plan actually supports that conclusion.
For some questions, one unit is adequate. For others, it isn’t. Identity testing on incoming raw material needs a different approach than assay testing on finished peptide vials. A label check needs a different approach than a potency, contaminant, or microbial assessment. The right plan always starts with the decision you actually need to make.
What FDA expects depends on your product category
For drug manufacturers, sampling sits inside the CGMP quality system. Components, drug product containers, and closures must be held from use until sampled, tested or examined as appropriate, and released by the quality control unit. Representative samples of each shipment of each lot must be collected, and the number of containers sampled should be based on appropriate criteria such as component variability, confidence level, supplier history, precision desired, and the amount needed for analysis and reserve.
For finished drug products, the sample plan has to support the release decision rather than simply generate a passing laboratory result. FDA’s CGMP regulation requires appropriate laboratory determination of satisfactory conformance to final specifications before release, and any sampling and testing plans must be described in written procedures that include the sampling method and number of units per batch to be tested.
For dietary supplements, 21 CFR 111.80 is very direct: firms must collect representative samples of components, packaging, labels, in-process materials, finished batches selected through a sound statistical sampling plan or every finished batch, received product for packaging or labeling, and packaged-and-labeled dietary supplements.
For food manufacturers, the preventive-controls rule is hazard-based. Product testing is not a fixed-number requirement for every food, but when it is used as a verification activity, the written procedures must be scientifically valid and identify what is being sampled, the relationship to specific lots, the number of samples, sampling frequency, test method, laboratory, and corrective-action process (21 CFR 117.165).
For food importers under the Foreign Supplier Verification Program, verification can include onsite audits, sampling and testing, supplier record review, or other appropriate activities (21 CFR 1.506). When sampling and testing are used, importers must retain documentation showing what was tested, lot information as appropriate, number of samples, test methods, dates, results, corrective actions, laboratory identity, and qualified-individual documentation.
Research peptides: the reseller challenge
Distributors and resellers often didn’t manufacture the material they sell. You may receive finished vials, bulk powder, or labeled product with a supplier COA but limited visibility into synthesis, purification, filling, lyophilization, cleaning records, packaging controls, or line clearance.
That doesn’t make testing less important. It makes thoughtful sampling more important.
A single vial from a shipment can tell you something useful about that vial. But if the shipment has multiple boxes, sublots, fill positions, or packaging groups under one lot number, one vial rarely tells the whole story. We have seen anonymized peptide projects where multiple vials from the same shipment did not behave the same way: most units looked acceptable, while one unit from a different box showed a meaningful fill or assay difference that a single random vial, or a composite, could have missed.
This is especially true when it comes to sterility testing. Microbial contamination is often intermittent and may only show up in products that use one specific fill head, or that were processed at the beginning or end of the batch. Sterility assurance cannot be established by testing more finished units alone; it depends on validated process controls, an appropriate sampling plan, a suitable method, and the intended decision.
A practical peptide sampling plan should document the supplier lot, received quantity, number of containers, number of units selected, selection method, whether units were tested individually or composited, storage conditions, and the decision rule if something fails.
For identity or gross purity screening, fewer units may be reasonable. For vial strength, fill consistency, residual solvents, water content, counterion, endotoxin, or sterility-related claims, a broader approach usually becomes necessary. When you sell by vial, individual-vial testing is almost always more informative than a composite.
A “research use only” label may be relevant, but it is not the whole analysis. Under 21 CFR 201.128, objective intent may be shown by labeling claims, advertising, oral or written statements, product design or composition, and the circumstances surrounding distribution. Regardless of regulatory category, customers still rely on the quality data you provide.
Manufacturers and distributors have different advantages
Manufacturers can tie samples to process knowledge: blend time, fill order, lyophilizer shelf, equipment train, packaging lane, hold time, cleaning sequence, or deviation history. A manufacturer can deliberately sample beginning, middle, and end of fill; multiple drums or totes; different packaging lanes; or different in-process control points.
Distributors usually can’t do that. Their plan has to rely on shipment mapping, cross-container selection, supplier COA comparison, independent testing, chain of custody, and reserve samples. If you received 20 boxes, pulling three vials from the same box is weaker than pulling across the shipment. If a supplier COA says the lot passed, independent testing from multiple received locations can help verify whether the material you actually received is consistent with that COA.
In both cases, the lab report becomes far more valuable when it includes the story of where the sample came from. “One vial tested” is a thin record. “Three vials from three separate cartons across the shipment, tested individually” is a record that stands up better in a customer review, supplier dispute, or internal quality file.
Where AQL fits, and where it doesn’t
Standards like ISO 2859-1:2026 are useful for lot-by-lot inspection by attributes using sampling schemes indexed by acceptance quality limit, or AQL. ISO describes this as a system of acceptance sampling plans for inspection by attributes, used to assess product or process quality through sample-based inspection.
AQL plans are useful, but they are not magic. They don’t prove a lot is perfect. They give you a statistical decision framework based on sample size, lot size, inspection level, and allowable defects.
Use them where they fit: visual defects, label checks, packaging counts, damaged units, missing units, or other attribute-type inspections. Pair them with product-risk assessment for potent actives, microbiological hazards, cross-contamination, adulterants, or high-consequence contaminants. AQL can support a sampling rationale. It should not replace scientific judgment.
Composite testing: helpful, or hiding the problem?
Compositing can save time and money when you only need an average for a well-mixed bulk material or a broad lot-level screen. It can be reasonable for certain ingredient checks, contaminant screens, or low-risk monitoring programs.
But compositing can also hide the problem. As an example from 21 CFR 211.84(c)(4), where location-based samples are needed to detect a nonconforming subdivision, top, middle, and bottom samples may not be composited.
One underfilled peptide vial can disappear into the average. A localized contaminant can get diluted. A mislabeled subgroup of product may not be caught by chemistry at all. A blend uniformity problem may be masked if the composite combines high and low portions of the lot.
The right question is not simply, “Can we composite?” The right question is, “Will compositing still answer the quality question we care about?”
A simple 7-question checklist we run with clients

Before pulling samples, run through these questions. They take a few minutes and prevent a surprising number of later problems.
- What population are you deciding about: one unit, one shipment, one supplier lot, one manufacturing batch, one pallet, one drum, or one packaging run?
- What exactly are you trying to prove: identity, assay, purity, fill consistency, label accuracy, microbial quality, heavy metals, residual solvents, pesticides, mycotoxins, allergens, adulterants, or something else?
- How high is the risk: new supplier, high-potency peptide, difficult matrix, customer complaint, import verification, supplier qualification, regulatory release, or routine monitoring?
- Are you sampling from the spots most likely to show variation: beginning/middle/end, different cartons, different pallets, different containers, different lyophilizer shelves, different packaging positions, or different received cases?
- Should the units be tested individually, composited, or both?
- What is your predefined decision rule if one unit fails, the composite fails, results disagree with the supplier COA, or the value sits near the specification limit?
- Are you keeping records such as chain of custody, photos, sampling notes, supplier COA, storage conditions, and unopened reserve samples that will hold up in an audit, customer review, or supplier discussion?
Answering these questions turns sampling from guesswork into a defensible quality decision.
What to send with your samples
The sample itself matters. The context matters just as much. When you submit samples to Vanguard Laboratory, include as much of the following as you can:
- Product details: Product name, CAS number, supplier, lot or batch number, received quantity, and number of containers or vials.
- Sampling context: Sample selection method and whether the sample represents one unit, one container, one shipment, one supplier lot, or the full lot.
- Test requirements: Requested tests, your specifications or acceptance criteria, and the supplier COA.
- Manufacturing records: Manufacturing stage, container number, fill position, packaging run, in-process location, retained-sample status, or deviation reference.
- Distribution records: The specific box, carton, case, pallet, or shipment segment the sample came from, plus any unopened reserves you kept.
This extra context lets the report answer the question you usually care about most: “What does this result actually tell me about the product I’m about to release, sell, use, or investigate?”
Bottom line
Sampling is the bridge between the physical product and the data reported.
Get it right and testing can help detect potency variation, contamination, supplier issues, labeling errors, degradation, or shipment problems before they reach customers. Get it wrong and even a perfect lab result can create false confidence.
We see this every week at Vanguard. That’s why we do more than run the test. We help clients select appropriate tests, define practical sampling plans, and interpret results in the context of product risk, regulatory category, and business need.
Send us your product type, lot size, supplier context, and release decision before you pull samples. We can recommend what to submit so the report is more likely to answer the question that matters.
It takes a few minutes up front and can save weeks of uncertainty later.
Dustin Newman is the owner and head laboratory technician at Vanguard Laboratory. He works directly with manufacturers, distributors, and resellers to design practical, risk-appropriate sampling and testing plans.